
Most people know that viruses are notoriously tricky disease-causing pathogens to tackle. Unlike bacteria which are completely separate organisms, viruses are parasites which use a host cell’s own DNA-and-RNA-and-protein producing mechanisms to reproduce. As a result, most viruses are extremely small, as they need to find a way into a cell to hijack the cell’s machinery, and, in fact, are oftentimes too small for light microscopes to see as beams of light have wavelengths that are too large to resolve them.
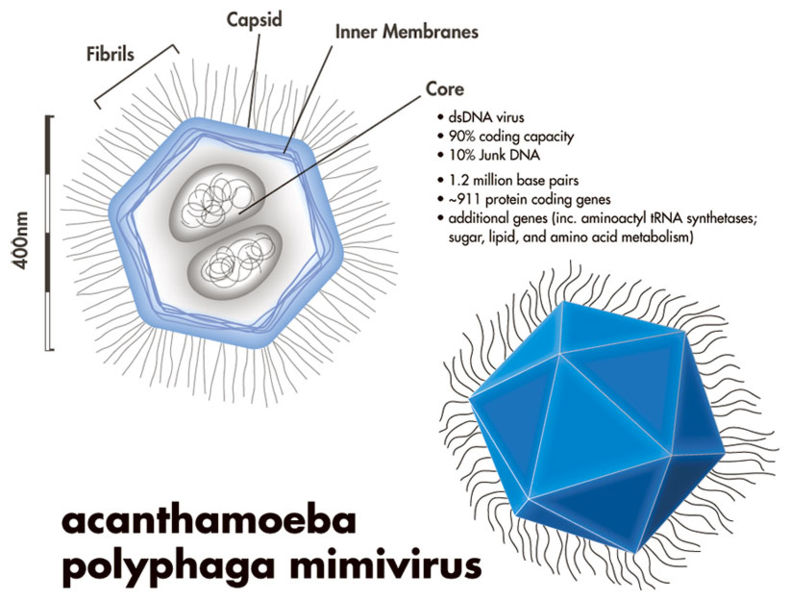
However, just because most viruses are small, doesn’t mean all viruses are. In fact, giant Mimiviruses, Mamaviruses, and Marseillesviruses have been found which are larger than many bacteria. The Mimivirus (pictured below), for instance, was so large it was actually identified incorrectly as a bacteria at first glance!
{kind=link}
Little concrete detail is known about these giant viruses, and there has been some debate about whether or not these viruses constitute a new “kingdom” of life (the way that bacteria and archaebacteria are), but one thing these megaviruses have in common is that they are all found within amoeba!
This month’s paper (HT: Anthony) looks into the genome of the Marseillesvirus to try to get a better understanding of the genetic origins of these giant viruses. The left-hand-side panel of picture below is an electron micrograph of an amoeba phagocytosing Marseillesvirus (amoeba, in the search for food, will engulf almost anything smaller than they are) and the right-hand-side panel shows the virus creating viral factories (“VF”, the very dark dots) within the amoeba’s cytoplasm. If you were to zoom in even further, you’d be able to see viral particles in different stages of viral assembly!
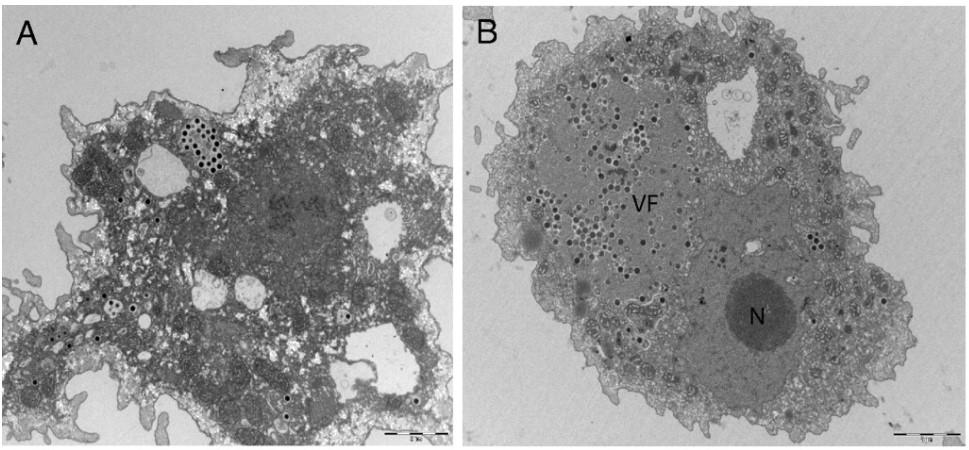
Ok, so we can see them. But just what makes them so big? What the heck is inside? Well, because you asked so nicely:
- ~368000-base pairs of DNA
- This constitutes an estimated 457 genes
- This is much larger than the ~5000 base pair genome of SV40, a popular lab virus, the ~10000 base pairs in HIV, the ~49000 in lambda phage (another scientifically famous lab virus), but is comparable to the genome sizes of some of the smaller bacterium
- This is smaller than the ~1 million-base pair genome of the Mimivirus, the ~4.6 million of E. coli and the ~3.2 billion in humans
- 49 proteins were identified in the viral particles, including:
- Structural proteins
- Transcription factors (helps regulate gene activity)
- Protein kinases (primarily found in eukaryotic cells because they play a major role in cellular signaling networks)
- Glutaredoxins and thioredoxins (usually only found in plant and bacterial cells to help fight off chemical stressors)
- Ubiquitin system proteins (primarily in eukaryotic cells as they control which proteins are sent to a cell’s “garbage collector”)
- Histone-like proteins (primarily in eukaryotic cells to pack a cell’s DNA into the nucelus)
As you can see, there are a whole lot of proteins which you would only expect to see in a “full-fledged” cell, not a virus. This begs the question, why do these giant viruses have so many extra genes and proteins that you wouldn’t have expected?
To answer this, the researchers ran a genetic analysis on the Marseillesvirus’s DNA, trying to identify not only which proteins were encoded in the DNA but also where those protein-encoding genes seem to come from (by identifying which species has the most similar gene structure). A high-level overview of the results of the analysis is shown in the circular map below:
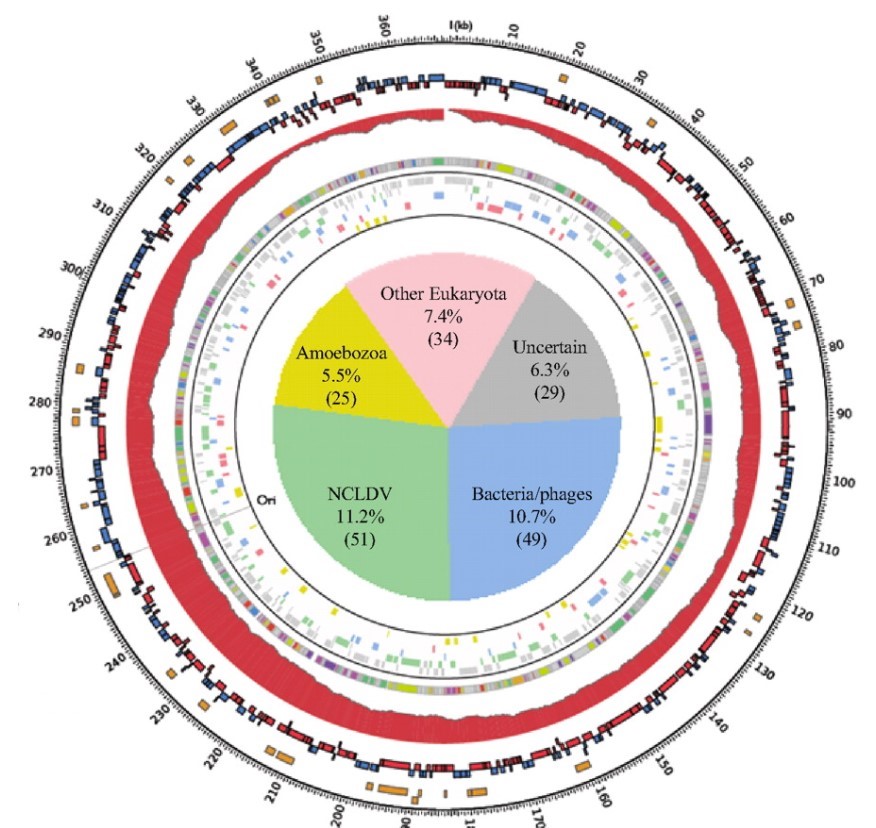


The outermost orange bands in the circle correspond to the proteins that were identified in the virus itself using mass spectrometry. The second row of red and blue bands represents protein-coding genes that are predicted to exist (but have yet to be detected in the virus; its possible they don’t make up the virus’s “body” and are only made while inside the amoeba, or even that they are not expressed at all). The gray ring with colored bands represents the researchers’ best guess as to what a predicted protein-coding gene codes for (based on seeing if the gene sequence is similar to other known proteins; the legend is below-right) whereas the colored bands just outside of the central pie chart represents a computer’s best determination of what species the gene seems to have come from (based on seeing if the gene sequence is similar to/the same as another species).
Of the 188 genes that a computational database identified as matching a previously characterized gene (~40% of all the predicted protein-coding genes), at least 108 come from sources outside of the giant viruses “evolutionary family”. The sources of these “misplaced” genes include bacteria, bacteria-infecting viruses called bacteriophages, amoeba, and even other eukaryotes! In other words, these giant viruses were genetic chimeras, mixed with DNA from all sorts of creatures in a way that you’d normally only expect in a genetically modified organism.
As many viruses are known to be able to “borrow” DNA from their hosts and from other viruses (a process called horizontal gene transfer), the researchers concluded that, like the immigrant’s conception of the United States of America, amoebas are giant genetic melting pots where genetic “immigrants” like bacteria and viruses comingle and share DNA (pictured below). In the case of the ancestors to the giant viruses, this resulted in viruses which kept gaining more and more genetic material from their amoeboid hosts and the abundance of bacterial and virus parasites living within.
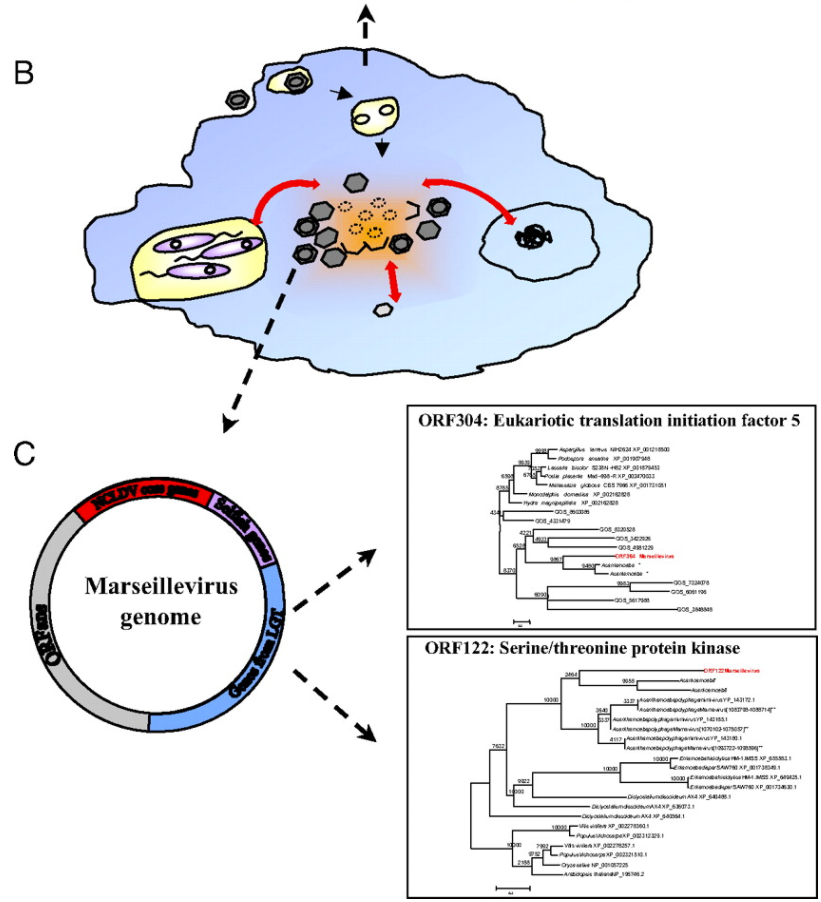
his finding is very interesting, as it suggests that amoeba may have played a crucial role in the early evolution of life. In the same way that a cultural “melting pot” like the US allows the combination of ideas from different cultures and walks of life, early amoeba “melting pots” may have helped kickstart evolutionary jumps by letting eukaryotes, bacteria, and viruses to co-exist and share DNA far more rapidly than “regular” natural selection could allow.
Of course, the flip side of this is that amoeba could also very well be allowing super-viruses and super-bacteria to breed…
Paper: Boyer, Mickael et al. “Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms.” PNAS 106, 21848-21853 (22 Dec 2009) – doi:10.1073/pnas.0911354106